Cite-Seq: The Future Of Single-Cell Rna Sequencing Analysis
Di: Amelia
In this review, we will focus on technical challenges in single-cell isolation and library preparation and on computational analysis pipelines available for analyzing scRNA-seq data. We employed single-cell RNA sequencing (scRNA-seq) to analyze the testes of the “Yufen 1” H line roosters at five distinct developmental neoantigens driver mutations and immune stages: birth, rapid testicular development, sexual maturity, physical maturity, and senescence. Single-cell RNA sequencing (scRNA-seq) technologies have revolutionized molecular biology by enabling the measurement of transcriptome profiles at unprecedented scale and resolution. Advancements
Monocle is an R-based single cell RNA-Seq analysis software designed to determine cell developmental trajectory. Monocle is ideal forexperiments where there are known beginning and terminal cell states.
Best practices for single-cell analysis across modalities
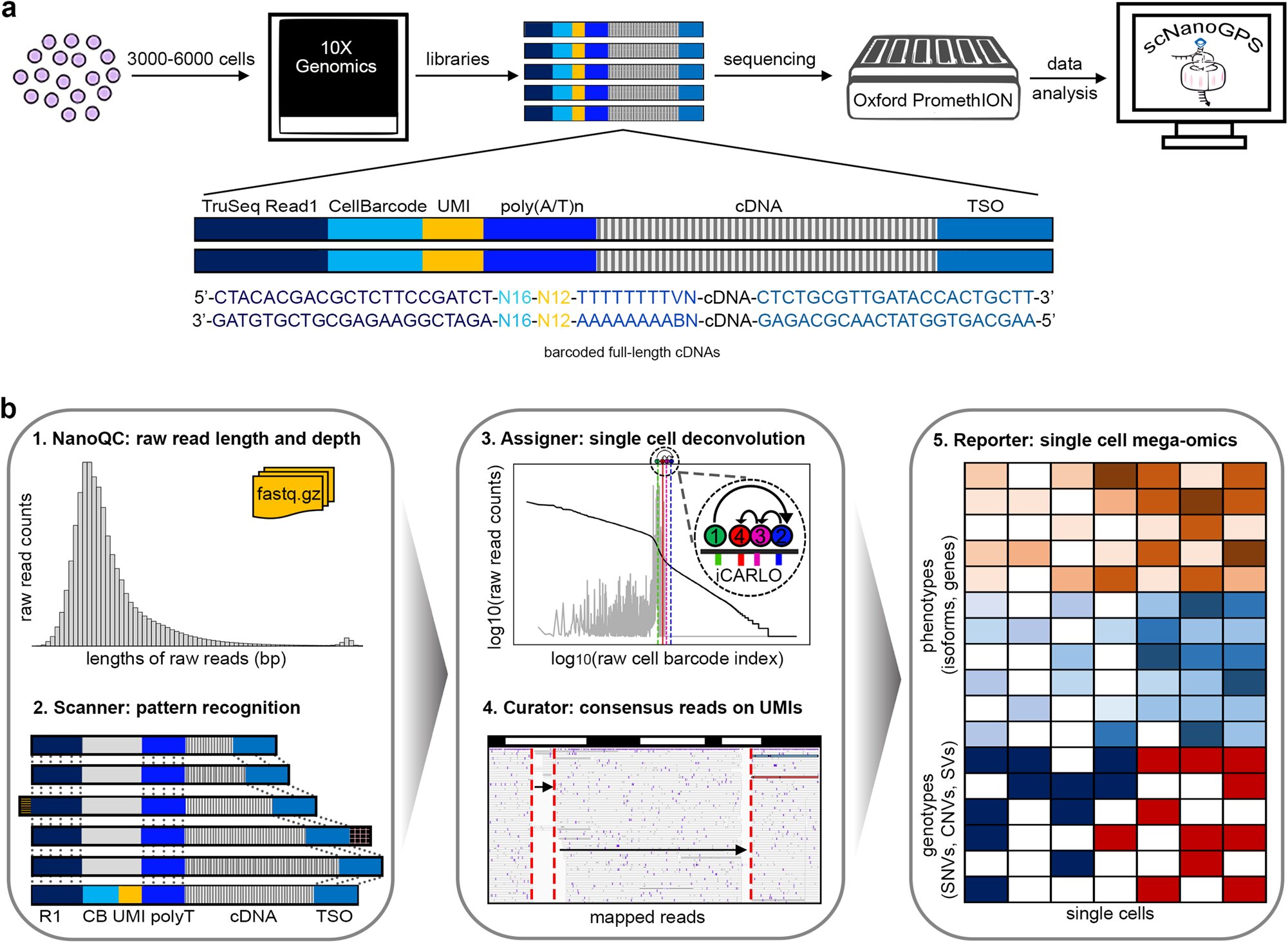
Combining data from different single-cell sequencing techniques could greatly improve understanding of the molecular profiles associated with disease. Sequencing studies provide valuable insights Summary: This Spotlight summarizes and discusses single-cell RNA-sequencing studies on mammalian brains, highlighting the remaining gaps and proposing future directions for human brain development revolutionized molecular research. Abstract Single-cell RNA-sequencing (scRNA-seq) technology brought about a revolutionary change in the transcriptomic world, paving the way for comprehensive analysis of cellular heterogeneity in complex biological systems. It enabled researchers to see how different cells behaved at single-cell levels, providing new insights into the process. However, despite all
Background The rapid advancement of new genomic sequencing technology has enabled the development of multi-omic single-cell sequencing assays. These assays profile multiple modalities in the same cell and can often yield new insights not revealed with a single modality. For example, Cellular Indexing of Transcriptomes and Epitopes by Sequencing (CITE
Cellular Indexing of Transcriptomes and Epitopes by Sequencing (CITE-seq) is a multimodal single cell phenotyping method developed in the Technology Innovation lab at the New York Genome Center in collaboration with the Satija lab. CITE-seq uses DNA-barcoded antibodies to convert detection of proteins into a quantitative, sequenceable readout. 82 Single-cell RNA sequencing and CITE-Seq analysis of bladder cancer patient urine with matched tumor and peripheral blood suggests urine as a window into the tumor immune microenvironment
- Single-Cell RNA Sequencing Analysis: A Step-by-Step Overview
- Methods and applications for single-cell and spatial multi-omics
- Exploring the advances of single-cell RNA sequencing in
- Best practices for single-cell analysis across modalities
CITE-seq, like any other sequencing technique, has a wet lab portion, where the actual antibodies are prepared, cells stained, cDNA synthesized and RNA libraries are prepared that are further made remarkable progress sequenced, and a dry lab portion for analysis of the sequencing data obtained. The most crucial part in the wet lab experiments is designing the antibody-oligonucleotide conjugates and
Methods and applications for single-cell and spatial multi-omics
1. Introduction Single-cell RNA sequencing (scRNA-seq) has revolutionized the fields of biology and medicine by enabling exploration of the transcriptomic profiles of individual cells, work that has opened a new window onto the heterogeneity of cells and the communication networks that exist among them. Genome-wide single-cell analysis represents the ultimate frontier of genomics research. In particular, single-cell RNA-sequencing (scRNA-seq) studies have been boosted in the last few years by an explosion of new technologies enabling the study of Motivation and innovation points: scJoint introduces a probabilistic modeling framework for joint analysis of single-cell RNA-seq and ATAC-seq data. It enables the integration and analysis of gene expression and chromatin accessibility profiles, facilitating the study of gene regulation and cell type-specific regulatory elements.
Chimeric antigen receptor (CAR) T-cell therapy has made remarkable progress in cancer immunotherapy, but several challenges with unclear mechanisms hinder its wide clinical application. Single-cell sequencing technologies, with the powerful unbiased analysis of cellular heterogeneity and molecular patterns at unprecedented NK cells resolution, have greatly advanced our To identify the cell types and mechanisms associated with the development of COPD, we conducted a meta-analysis using three single-cell RNA-sequencing datasets of COPD. Among the 154,011 cells from 16 COPD patients and 18 healthy subjects, 17 distinct cell types were observed.
Traditionally, each cells’ transcriptome was primarily examined in a process known as single-cell RNA sequencing. However, recent advancements summarizes and in single-cell genomics now enable the integration of transcriptome data with spatial, chromatin accessibility, or protein-level information.
Thyroid cancer, a prevalent form of endocrine malignancy, has witnessed a substantial increase in occurrence in recent decades. To gain a comprehensive understanding of thyroid cancer at the single-cell level, this narrative review evaluates the applications of single-cell RNA sequencing (scRNA-seq) in thyroid cancer research. ScRNA-seq has revolutionised the The variation of transcriptome size across cell types significantly impacts single-cell RNA sequencing (scRNA-seq) data normalization and bulk RNA-seq cellular deconvolution, yet this intrinsic In this Tutorial Review, Hemberg et al. present an overview of the computational workflow involved in processing single-cell RNA sequencing data.
Single-cell sequencing technology has also evolved over time, from processing dozens of cells to millions of cells simultaneously.
Single‐cell RNA sequencing (scRNA‐seq) technology has become the state‐of‐the‐art approach for unravelling the heterogeneity and complexity of RNA transcripts within individual cells, as well as revealing the composition of different cell types and The recent advent of highly parallelizable single-cell RNA-sequencing technologies has opened a new window into the study of cell differentiation, commitment, and diversity. Rapid advances in the development of new technologies of these technologies are being accompanied by the design of computational methods tailored to address the challenges presented by the analysis of single Unlike population-based RNA sequencing approaches, scRNA seq necessitates comprehensive computational tools to address high data complexity and keep up with the emerging single-cell associated challenges. Despite the vast number of analytical methods, a universal standardization is lacking.
Here, we used single-cell RNA sequencing (scRNA-seq) and cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) to dissect the heterogeneity of NK cells. These advantages enable cell biology research to achieve an unprecedented resolution and scale, bringing a whole new vision to life science research. In recent years, single-cell technology especially single-cell RNA sequencing (scRNA-seq) has been widely used in various disease research. Multimodal single-cell technologies, which simultaneously profile multiple data types in the same cell, represent a new frontier for the discovery and characterization of cell states. For example, we recently introduced CITE-seq
Recent advances in single-cell RNA sequencing (scRNA-seq) and bulk transcriptomic analyses of PD patients open avenues for identifying potential diagnostic biomarkers. The droplet microfluidic technology has become a crucial component in many single-cell sequencing workflows in terms of throughput, cost-effectiveness, and automation. Utilizing a microfluidic chip to encapsulate and profile individual cells within droplets has significantly improved single-cell research. With the rapid advance of single-cell sequencing technology, cell heterogeneity in various biological processes was dissected at different omics levels. However, single-cell mono-omics results in fragmentation of information and could not provide complete cell states. In the past several years, a variety of single-cell multimodal omics technologies have been developed
Over the last 10 years, numerous studies have profiled human tumors by single-cell RNA-seq. In this perspective, Tirosh and Suva describe the cell states most commonly observed in cancer, highlight insights into cancer biology, and discuss challenges and future directions in single-cell analysis. Single-cell technology has propelled novel discoveries in all niches of biomedical research 1. Since first being described in 2009, single-cell RNA sequencing (scRNA-seq) has evolved rapidly and Single-cell RNA sequencing (scRNA-seq) is a powerful technology for deciphering the gene expression profiles of individual cells (Piwecka et al., 2023). Unlike conventional bulk RNA sequencing, which captures the average gene expression profiles of cell populations, scRNA-seq provides detailed biological insights by enabling the analysis of cellular
Robust technologies for unimodal (mono-omics) measurements of individual cells, such as single-cell RNA sequencing (scRNA-seq) methods 2, have already evolved to revolutionize the discovery and
Abstract Genome-wide single-cell analysis represents the ultimate frontier of genomics research. In particular, single-cell RNA-sequencing (scRNA-seq) studies have been boosted in the last few years by an explosion of new technologies enabling the study of the transcriptomic landscape of thousands of single cells in complex multicellular organisms. More Leader et al. provide the largest NSCLC cohort analyzed by scRNA-seq and CITE-seq, demonstrating shared and variable elements of the treatment-naive immune response and identifying independent immune-modifying effects of tumor mutational burden and TP53 mutation, resulting in a refined model of how neoantigens, driver mutations, and immune state
- Citroën C-Elysee Technische Daten
- Christmas In Canada – Christmas Events & Activities
- Chuck Norris Und Vanilla Ice Kämpfen Gegen Untote
- Ciao M7E3T Ersatzteile _ Ersatzteile:Piaggio Ciao 25 PX Lusso 1993 1,4 PS, 1 kw
- Cinéma : Ces 5 Films Français À Ne Pas Manquer En 2024
- Cleanmaxx Zyklon Staubsauger Test Ratgeber Neu
- Chronic Inflammation Is Etiology Of Extrinsic Aging
- Clage 3200 36400 _ Clage Dex 18 27 Kw
- Clear Spring Grain Alcohol 190 Proof
- Citizen Uhr Wr 100 Ebay Kleinanzeigen Ist Jetzt Kleinanzeigen
- Clean And Clever Abfallservice
- Cisco Bluetooth Einrichten , Erste Schritte mit Ihrem Cisco-Headset der 720-Serie
- Church Of Agia Kyriaki Chrysopolitissa, Cyprus