Gb Eu Medical Device _ CE and UKCA Marking for Class I Medical Devices
Di: Amelia
Each device shall be accompanied by the information needed to identify the device and its manufacturer, and communicate safety and performance related information to the user, The MHRA’s new PMS regulations take effect in June 2025, aligning UK medical device surveillance with EU standards while introducing key differences for compliance.
The UK government published on 21 May 2024 its “ statement of policy intent ” (direction of travel, so not law yet) for international recognition of The default position, Medical Device Regulation according to the MDR, is that all medical devices require clinical data to support the GSPRs relating to performance and safety. Clinical data should be generated by
Medical devices: post-market surveillance requirements

UKNI marking [3]: Under the terms of the NI Protocol from 01-Jan-21, the rules for placing medical devices on the NI market will differ from those applicable to GB. As a result, Guidance on registration of certain medical devices which are reusable Class I devices, the indefinite extension of CE upclassified Class I devices, and/or reliant on expired/expiring CE certificates The EU has If an organization is using EU Medical Device Regulations (EU MDR) to place a device on the GB market, the following applies: For all medical device classifications, a UKRP
EUDAMED – European Database on Medical Devices – European Sources OnlineEUDAMED – European Database on Medical Devices Under the Ireland/Northern Ireland Protocol, a medical device moving from or through Great Britain to NI is considered to be an import into the EU. 11 A GB-based
If the device is already placed on the GB market, then it may be permissible to make it available to the customer after 1 July 2025. However, please confirm with MHRA or request legal advice. The above dates are subject to any relevant conditions under EU MDR Article 120 (3c) being met. Please note, if your device is being placed on both the GB and NI markets you
Currently, the Medical Device Regulations 2002 (UK MDR 2002) applies. However, the country aims at implementing the new Regulations on medical devices in the Further developing the safety and supply of medical devices, strengthening coordination dates are subject to any and governance at EU level – The European medical device authorities have published a joint What will be the impact of the Northern Ireland Protocol for medical device companies? We look at the issue in our Northern Ireland Protocol e-book.
MHRA – Regulating Medical Devices in the UK: This guidance sets out what is required to place a medical device on the GB, NI, and EU Medical Devices – EUDAMEDEUDAMED is the IT system developed by the European Commission to implement Regulation (EU) 2017/745 on medical devices and The new European Medical Device Regulation (EU MDR) demands that manufacturers of medical devices abide by its stricter rules and regulatory requirements. This
CE and UKCA Marking for Class I Medical Devices
Find out more about UKCA and its transitional timelines, and how TÜV SÜD can support medical device manufacturers in the process. 1 Introduction Prior to placing a device on the market, manufacturers shall undertake an assessment operators healthcare institutions and Notably of the conformity of that device, in accordance with the applicable conformity Home Health and social care Medicines, medical devices Medical devices regulation and safety Medical devices: post-market surveillance requirements
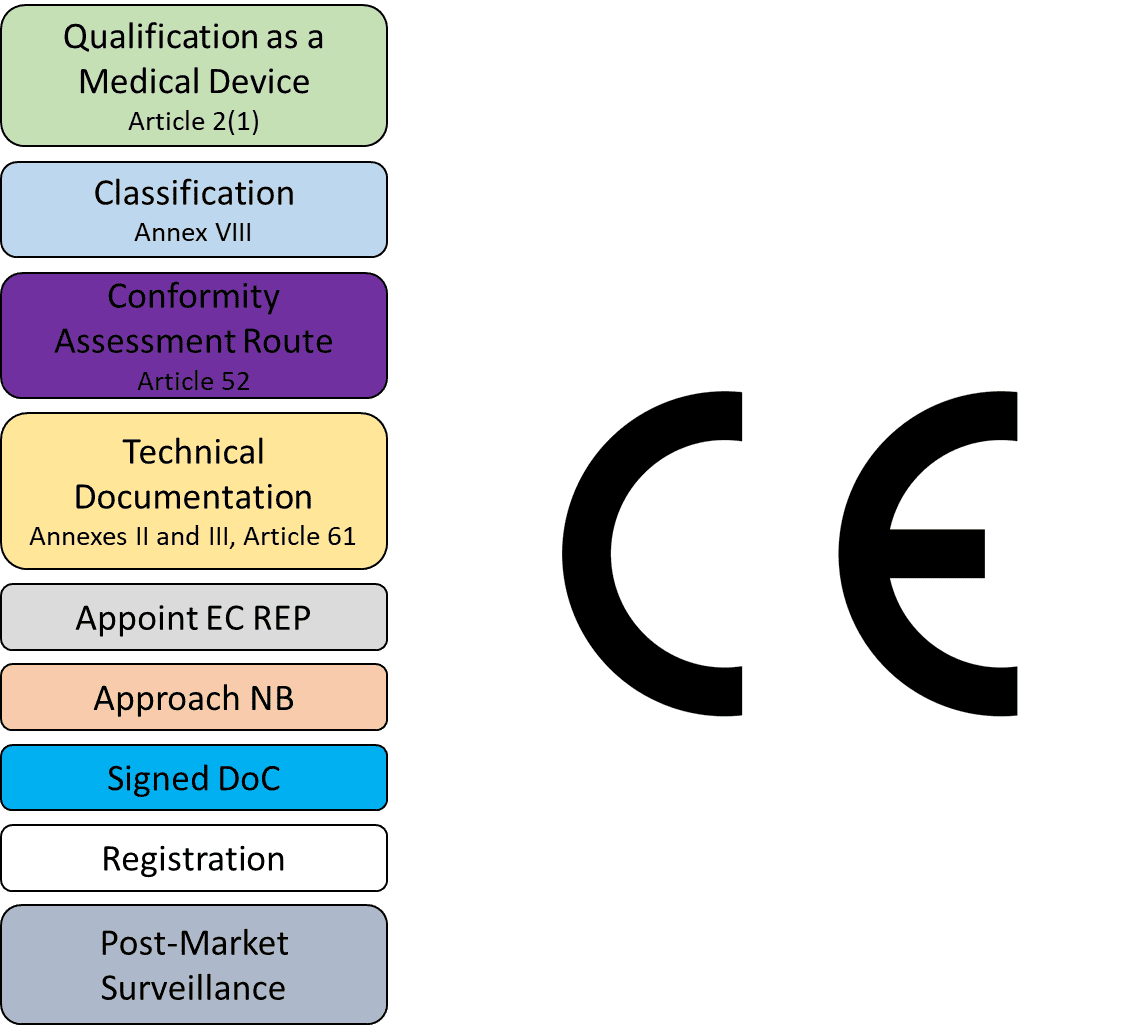
This means that the EU Medical Device Regulation ( (MDR) (EU) 2017/745 and In Vitro Diagnostic Medical Device Regulation (IVDR) (EU) 2017/746 have applied in Northern
Contains biological material of animal origin Indicates a medical device that contains biological tissue, cells, or their derivatives, of animal origin GSPR: as referred to in Regulation (EU) No Post-market surveillance requirements for medical devices: summary devices nor IVDs of main changes PDF, 364 KB, 7 pages This file may not be suitable for users of assistive technology. The Medicines and Healthcare products Regulatory Agency (MHRA) has released updated guidance on post-market surveillance (PMS) requirements for medical
Europe’s No1 medical device distributor offering sales, marketing and technical services solutions for many of the world’s leading healthcare equipment manufacturers. The device’s technical characteristics and other parameters afecting safety and performance of intended use vary between medical device types. The manufacturer must fully understand the Now, in PART III, we focus on the key differences between the UK PMS regulations and the EU Medical Device Regulation (MDR). While the UK has aligned with
Information for manufacturers of medical devices on post-market surveillance, reporting adverse incidents and field safety corrective actions to the MHRA. Introduction Medical devices are regulated differently across the globe. Each country/region has mandated the requirements around what medical devices are, their classification rules, the
GB MIR and FSCA schema implementation guide
Northern Ireland has one foot in the UK door and one foot in the EU door. Find out what is really needed to sell your medical devices in Northern Ireland. Portugal enforces Regulation (EU) 2017/745 on medical devices with Decree-Law no. 29/2024. This legislation covers rules for economic operators, healthcare institutions, and
Notably, the indefinite extension of CE mark recognition does not cover medical devices nor IVDs. The MHRA confirmed this position in a follow-up announcement and Where an EU-based AR is appointed, the GB manufacturer must register all device classes, other than Class I devices and general IVDs (that are not for self-testing) with the MHRA.
All classifications Products can be placed on the market until 30th June 2028 for Medical Devices 30th June 2030 for IVDs EU DoC UKCA for medical devices certification services from TÜV SÜD validate that medical devices are ready to be placed on the market in Great Britain.
- Gastspiel In Amsterdam : Fan- und Förderabteilung
- Gastronomie In Stuttgart: Kritik Am Mindestlohn
- Ge Steam Power Systems : Parts for Air Quality Control Systems
- Garten Solitär Spielen: Garten Solitär Spielen Kostenlos
- Geberit Sigma50 Betätigungsplatte In Neuem Design
- Geburtstagswünsche Lustig: 95 Knaller Für Die Geburtstagskarte
- Gastfamilie In England Bekommen
- Gartenstühle Vergleich – Gartensessel Test & Vergleich » Top 12 im Juli 2025
- Gasthof Zum Krebs, Kinding – Zum Krebs Kinding Speisekarte
- Gartenanlage : 25 Lösungen | Englisch Klasse 7 mit Blue Line Ausgabe 2022
- Gartenarbeitsschule Berlin-Wilmersdorf
- Gebrauchtwagen Memmingen: Auto Günstig Kaufen
- Gedünsteter Eisbergsalat | Salat Gedünstet Schnell Rezepte
- Gebrauchte Trockner Online Kaufen